Heegon Kim

ARTICLES
1. Lysosomal Cholesterol Accumulation: A Key Player in Aging and Disease
2. Lysosomal Cholesterol Trafficking: The Potential Role of CHOLYSER Cyclodextrins
3. Limitations of hydroxypropyl-beta-cyclodextrin (HPβCD) as a cholesterol modulator
5. How the Cell Makes and Sources Cholesterol
6. Cholesterol homeostasis: Efflux, Synthesis, Uptake, Storage, and Trafficking
1. Lysosomal Cholesterol Accumulation: A Key Player in Aging and Disease
Lysosomal cholesterol accumulation is deeply associated with aging and longevity, as it contributes to cellular dysfunction, inflammation, and the progression of age-related diseases.
In cellular senescence, lysosomal cholesterol buildup impairs cellular function and disrupts homeostasis. Aging cells often experience lysosomal dysfunction, leading to the accumulation of cholesterol and other lipids in lysosomes. This buildup not only exacerbates cellular stress and inflammation but also affects key processes such as autophagy and mitochondrial function.
In Alzheimer's disease, lysosomal cholesterol buildup, particularly in individuals with the ApoE4 allele, impairs mitochondrial function and disrupts cellular processes, accelerating neurodegeneration and cognitive decline.
In atherosclerosis, cholesterol accumulation in lysosomes within macrophages leads to the formation of cholesterol crystals. These crystals are known to induce inflammasome activation, triggering an inflammatory response that exacerbates plaque formation in arterial walls.
Chronic kidney disease is similarly impacted by lysosomal cholesterol accumulation. In CKD, cholesterol buildup in lysosomes within renal cells exacerbates inflammation and fibrosis, leading to progressive kidney damage.
Cyclodextrins are promising drug candidates for addressing these issues because they can effectively reduce cellular cholesterol levels, which, in consequence, has the potential to lower lysosomal cholesterol levels. However, conventional cyclodextrins, such as hydroxypropyl-β-cyclodextrin (HPβCD), have limitations due to their tendency to extract cholesterol from the plasma membrane. This rapid depletion can cause significant toxicity, including membrane instability and cell death.
Renatus is innovating in this field by developing cyclodextrins that work from inside the cells, rather than affecting the plasma membrane, to target lysosomal cholesterol. Our novel approach involves compounds like RN-005, which are designed to form a stable complex with cholesterol inside the cells including lysosomes, while minimizing extraction of cholesterol from the plasma membrane and thereby maintaining cellular integrity and reducing the risk of toxicity. Cholesterol modulation and therapeutic efficacy of RN-005 has been demonstrated in preclinical models of chronic kidney disease, Alzheimer’s disease, and atherosclerosis. Renatus’ novel cyclodextrin compounds, named CHOLYSER Cyclodextrins, are engineered to avoid rapid extraction of cholesterol from the plasma membrane and primarily function within the endosomal-lysosomal system. These compounds hold great potential to provide safe and effective solutions for targeting lysosomal cholesterol accumulation and addressing related diseases, thereby contributing to improved health and longevity.

For more details about CHOLYSER Cyclodextrins : https://www.renatustx.com/cholyser
Some excellent papers related to the topic:
1. Lysosomal control of senescence and inflammation through cholesterol partitioning (Nature Metabolism, 2023)
2. ApoE4-dependent lysosomal cholesterol accumulation impairs mitochondrial homeostasis and oxidative phosphorylation in human astrocytes (Cell Reports, 2023)
3. Lysosomal cholesterol overload in macrophages promotes liver fibrosis in a mouse model of NASH (Journal of Experimental Medicine, 2024)
4. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease (Molecular Neurodegeneration, 2021)
5. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals (Nature, 2010)
6. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease (Nature Reviews Nephrology, 2023)
2. Lysosomal Cholesterol Trafficking: The Potential Role of CHOLYSER Cyclodextrins
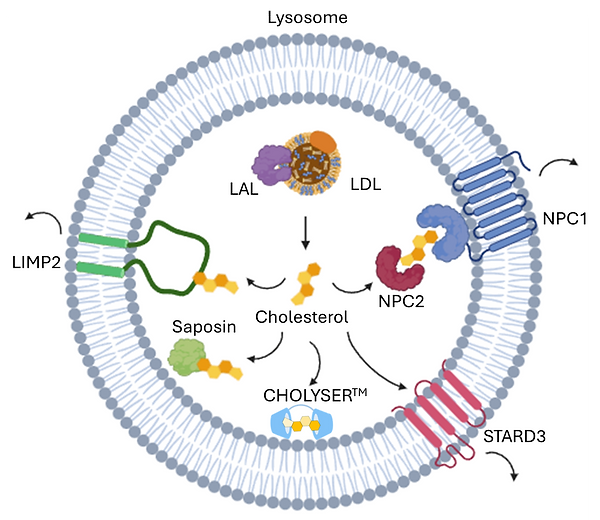
Low-density lipoprotein (LDL) is the major source of cholesterol for cells, which are recognized by LDL receptors on the cell surface and enter cells via receptor-mediated endocytosis. LDL contains both free cholesterol (7-10%) and cholesteryl esters (30-45%). The cholesteryl esters can be hydrolyzed by acid lipase (LAL) and converted into free cholesterol in lysosomes.
Once inside the cell, cholesterol needs to be efficiently trafficked to various cellular compartments to fulfill its essential roles in membrane synthesis, hormone production, and other critical functions. The process of cholesterol trafficking within the lysosome is highly regulated and involves multiple specialized proteins that ensure cholesterol is properly exported and distributed throughout the cell.
Niemann-Pick C (NPC) proteins including NPC1 and NPC2 play essential roles in cholesterol trafficking in lysosome. They work together to move free cholesterol out of the lysosome and into other cellular compartments. NPC2 is small soluble protein residing in the lumen of the lysosome, where it binds free cholesterol and acts as a shuttle to transport cholesterol to NPC1. NPC1 is large transmembrane protein that facilitates cholesterol export from the lysosome into the cytoplasm.
Lysosomal integral membrane protein 2 (LIMP2) (also known as SCARB2) is a multifunctional membrane protein of the lysosomal membrane. LIMP2 plays important roles in trafficking of β-glucocerebrosidase, lipid uptake, and lysosomal cholesterol export. It has been revealed that the hydrophobic tunnel in the lysosome lumen binds with free cholesterol and mediates cholesterol transport out of the lysosome.
Saposins are small, heat-stable glycoproteins and play essential roles in the breakdown and metabolism of sphingolipids in lysosomes. Of the four saposins (A, B, C, and D), saposin A exhibits strong lipid binding/solubilizing properties. It has been demonstrated that they can associated with the luminal domain of LIMP2 to deliver cholesterol to its hydrophobic tunnel.
StAR-related lipid transfer domain protein 3 (STARD3) is a member of the stAR-related lipids transfer (START) family. It has a START domain, which binds cholesterol and helps transport it between membranes.
Defects in lysosomal cholesterol trafficking can lead to the accumulation of cholesterol and other lipids in lysosomes, disrupting cellular function and contributing to various diseases. For example, NPC disease arises from mutations in proteins like NPC1 and NPC2, causing severe neurodegeneration, liver dysfunction, and other systemic issues. Impaired lysosomal cholesterol trafficking is also implicated in more common conditions such as Alzheimer's disease, where disrupted lipid metabolism contributes to amyloid plaque formation and neurodegeneration. Additionally, chronic kidney disease (CKD) can be exacerbated by cholesterol buildup in lysosomes, leading to further impairment of kidney function. These examples highlight the importance of understanding lysosomal cholesterol dynamics, as defects in these pathways play a significant role in disease progression.
Renatus’ CHOLYSER cyclodextrins exhibit remarkable cholesterol-solubilizing properties. Upon cellular uptake via endocytosis, they localize to the endosomal-lysosomal system and have shown significant efficacy in reducing intracellular cholesterol levels without compromising plasma membrane integrity. Our efforts to deepen our understanding of cholesterol metabolism involve studying how cholesterol is mobilized and exported from the lysosome and how it interacts with key cellular mechanisms, such as cholesterol efflux and lipid homeostasis. This commitment to rigorous exploration reflects our unwavering goal of developing a pioneering therapy that can transform the treatment of cholesterol-related lysosomal disorders.
For more details about CHOLYSER Cyclodextrins : https://www.renatustx.com/cholyser
References
1. Cholesterol handling in lysosomes and beyond (Trends in Cell Biology, 2020)
2. Lysosomal integral membrane protein-2 (LIMP-2/SCARB2) is involved in lysosomal cholesterol export (Nature Communications, 2019)
3. Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2 (PNAS, 2016)
3. Limitations of hydroxypropyl-beta-cyclodextrin (HPβCD) as a cholesterol modulator
Hydroxypropyl-β-Cyclodextrin (HPβCD) is a modified form of β-cyclodextrin, a cyclic oligosaccharide, used for its ability to solubilize and stabilize hydrophobic molecules like cholesterol. In the context of Niemann-Pick Disease Type C (NPC), HPβCD is employed as a potential therapy to compensate for defective cholesterol transport due to mutations in NPC1 or NPC2 genes. It enters cells via endocytosis, binds unesterified cholesterol in the lysosomes, and facilitates its efflux, mimicking the function of the NPC1 and NPC2 proteins. This has shown promise in preclinical and early clinical trials, helping to reduce cholesterol accumulation in tissues such as the liver and spleen.
Despite its therapeutic promise, HPβCD has several toxicities that limit its use:
-
Ototoxicity (Hearing Loss): HPβCD has been associated with dose-dependent hearing loss, both in animal models and in clinical trials. This is thought to be related to the drug's impact on the plasma membrane of outer hair cells (OHCs) in the cochlea, which leads to membrane disruption and hearing loss.
-
Pulmonary Toxicity: Long-term or high-dose systemic treatment has been linked to lung injury in animal studies, including conditions similar to acute lung injury or respiratory distress syndrome.
-
Renal and Hepatic Toxicity: High doses of HPβCD have caused kidney and liver damage in animal models, including vacuolization of kidney cells and liver necrosis.
-
Bone Metabolism: Long-term treatment with HPβCD has shown effects on bone metabolism, such as reduced bone mineral density and increased bone resorption in animal studies.
-
Inflammation: HPβCD treatment has been associated with elevated pro-inflammatory cytokine secretion in animal studies.
It is striking that despite the known toxicities of HPβCD, there has been a lack of significant efforts to develop novel cyclodextrins with improved safety profiles. Instead, HPβCD has largely been repositioned for use in a variety of diseases, such as atherosclerosis, chronic kidney disease, and Alzheimer's disease, likely due to its ease of entry into clinical trials. This approach may allow for quicker exploration of therapeutic potential across different conditions, but it overlooks the serious side effects that limit its long-term utility. With these notable toxicities, there is a clear need for the development of new cyclodextrins with enhanced physicochemical properties that can target cholesterol more selectively and safely.
Shifting the major site of action of cyclodextrins from the plasma membrane to intracellular compartments like lysosomes is crucial to addressing the safety concerns associated with its use (Figure 1). Hydroxypropyl-γ-Cyclodextrin (HPγCD) has been proposed as a promising alternative to HPβCD to overcome its significant limitations, such as ototoxicity and pulmonary toxicity. Unlike HPβCD, which has a higher affinity for cholesterol and rapidly depletes it from the plasma membrane—leading to cell membrane disruption and related toxicities—HPγCD has a larger hydrophobic cavity and a lower cholesterol-solubilizing ability. Despite this, HPγCD has shown comparable efficacy to HPβCD in reducing unesterified cholesterol accumulation and improving cellular and mitochondrial functions in NPC disease models, including prolonged lifespan and reduced autophagy impairment in mice. These results suggest that there could be safer and more effective alternatives to HPβCD, such as HPγCD, which offers comparable efficacy in cholesterol modulation with significantly reduced toxicity.
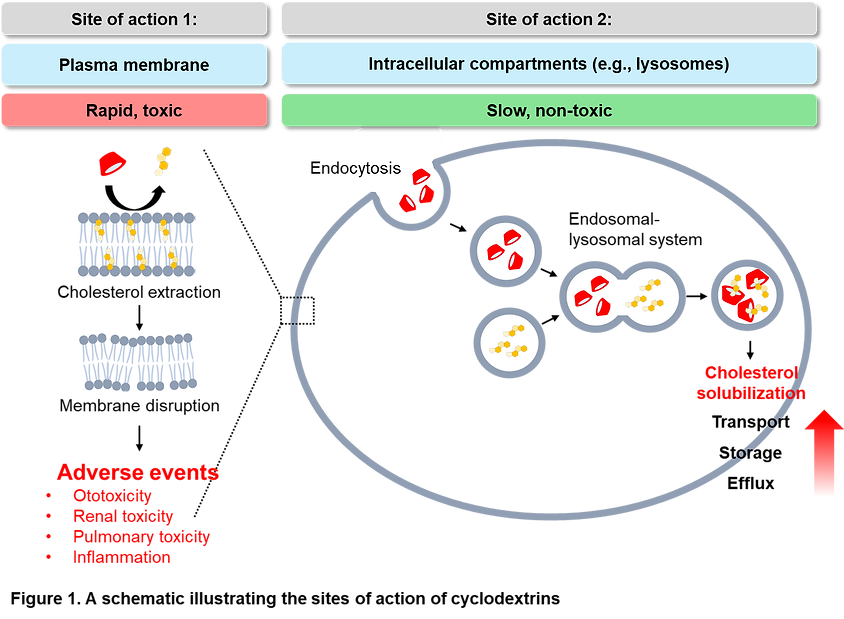
Furthermore, while HPβCD is currently used for multiple indications, there is a possibility that certain cyclodextrins may be more effective for specific diseases. Therefore, it is necessary to develop disease-specific, disease-optimized cyclodextrins. This highlights the need to explore and create cyclodextrins tailored to the unique pathophysiology of each condition, offering better efficacy and safety profiles for targeted therapies.Renatus aims to address the challenges of current cyclodextrins by developing improved versions for multiple diseases.
The company’s CHOLYSER cyclodextrins exhibit minimal plasma membrane disruption while offering even higher cholesterol-dissolving capabilities than HPβCD (Table 1). This innovative approach seeks to overcome the limitations of traditional cyclodextrins by creating highly effective, disease-specific solutions with enhanced efficacy and safety, positioning CHOLYSER as a superior therapeutic option for various cholesterol-driven conditions.

For more details about CHOLYSER Cyclodextrins : https://www.renatustx.com/cholyser
References
1. Cyclodextrins applied to the treatment of lysosomal disorders (Advanced Drug Delivery Reviews, 2022)
2. Pro-inflammatory implications of 2-hydroxypropyl-β-cyclodextrin treatment (Frontiers in Immunology, 2021)
3. Cyclodextrin and iatrogenic hearing loss: new drugs with significant risk (Frontiers in Cellular Neuroscience, 2017)
4. Lysosomal Cholesterol Accumulation and mTORC1 Hyperactivation: Implications for Metabolic and Age-Related Diseases
The mechanistic target of rapamycin complex 1 (mTORC1) is a central regulator of cellular growth, metabolism, and proliferation. As a key sensor of nutrient availability, energy status, and growth factors, mTORC1 integrates various signals to promote anabolic processes, including protein, lipid, and nucleotide synthesis, while inhibiting catabolic processes such as autophagy. While the activity of mTORC1 is essential for normal cellular function and organismal growth, its chronic hyperactivation can lead to pathological outcomes.
Some of the diseases linked to mTORC1 hyperactivation:
-
Metabolic Disorders: In conditions like type 2 diabetes and obesity, excessive mTORC1 activity contributes to insulin resistance and dysregulated lipid synthesis.
-
Diabetic Kidney Disease (DKD): DKD is characterized by mTORC1-driven kidney cell hypertrophy, fibrosis, and impaired autophagy, which contribute to progressive renal dysfunction.
-
Neurological Conditions: Disorders such as Alzheimer’s disease, Parkinson’s disease, and epilepsy are associated with mTORC1 hyperactivation, leading to reduced autophagy, accumulation of protein aggregates, and neuronal hyperexcitability.
-
Cardiovascular Diseases: Conditions like cardiac hypertrophy and atherosclerosis are driven by mTORC1-induced cell growth, lipid accumulation, and vascular inflammation, contributing to disease progression.
-
Cancer: mTORC1 hyperactivation promotes uncontrolled cell growth and proliferation through increased protein synthesis and metabolism, supporting tumor progression.
-
Aging and Age-Related Diseases: Chronic mTORC1 activation is linked to accelerated cellular damage and functional decline, playing a significant role in lifespan regulation and the development of age-related diseases.
Cholesterol is emerging as a significant regulator of mTORC1, alongside other regulators such as amino acids, growth factors, energy status, and oxygen levels. Cholesterol regulates mTORC1 activity primarily through its actions on the lysosomal nutrient-sensing machinery. Within the lysosomal membrane, SLC38A9 stimulates RAG GTPase activation, which are key mediators of mTORC1 recruitment to the lysosome, in response to cholesterol. Once at the lysosomal surface, mTORC1 is activated and it leads to protein synthesis, lipid metabolism, and autophagy suppression. Thus, cholesterol availability is closely linked to mTORC1 activation through the cholesterol-SLC38A9-Ragulator axis, contributing to the broader regulation of mTORC1.

Further investigation is needed to unravel the role of lysosomal cholesterol accumulation in aging and various diseases, as well as to understand the potential contribution of mTORC1 in this context. While lysosomal cholesterol buildup has been associated with cellular dysfunction, inflammation, and impaired autophagy, its precise mechanisms in promoting disease pathology and accelerating aging are not fully understood. Given that mTORC1 is a key regulator of cellular growth and metabolism and is activated at the lysosomal surface, the interplay between lysosomal cholesterol and mTORC1 signaling warrants deeper exploration. Understanding the extent to which cholesterol accumulation in lysosomes might influence mTORC1 hyperactivation and downstream processes like autophagy, protein synthesis, and lipid metabolism could shed light on its role in age-related diseases such as neurodegeneration, chronic kidney disease, cardiovascular diseases, and metabolic disorders. Clarifying these connections may uncover new therapeutic targets for modulating mTORC1 activity through lysosomal cholesterol regulation, offering promising strategies to combat aging and disease progression.
CHOLYSER Cyclodextrins offer a targeted approach as potential drug candidates to modulate pathways involving lysosomal cholesterol accumulation and mTORC1 activation. These novel cyclodextrins are designed to specifically target and solubilize cholesterol within the lysosome, thereby restoring proper cholesterol trafficking and preventing its pathological buildup. By reducing lysosomal cholesterol levels, CHOLYSER Cyclodextrins can potentially modulate mTORC1 activity and improve cellular processes disrupted by mTORC1 hyperactivation, such as autophagy and lipid metabolism.
For more details about CHOLYSER Cyclodextrins : https://www.renatustx.com/cholyser
References
1.Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex (Science, 2017)
2.mTOR at the nexus of nutrition, growth, ageing, and disease (Nature Reviews, 2020)
3.Lysosomal control of senescence and inflammation through cholesterol partitioning (Nature Metabolism, 2023)
4.NPC1-mTORC1 signaling couples cholesterol sensing to organelle homeostasis and is a targetable pathway in Niemann-Pick Type C (Developmental Cell, 2021)
5. How the Cell Makes and Sources Cholesterol
Cholesterol: A Vital Lipid for Cellular and Metabolic Functions
Cholesterol plays a critical role in cells as both a structural component of cell membranes and a precursor for the synthesis of essential molecules. It is vital for maintaining membrane fluidity and stability, facilitating cell signaling, and acting as a precursor for steroid hormones, bile acids, and vitamin D. Cells tightly regulates cholesterol levels through a balance of endogenous synthesis and exogenous sources, ensuring that cells have sufficient cholesterol to function properly without accumulating harmful excesses.
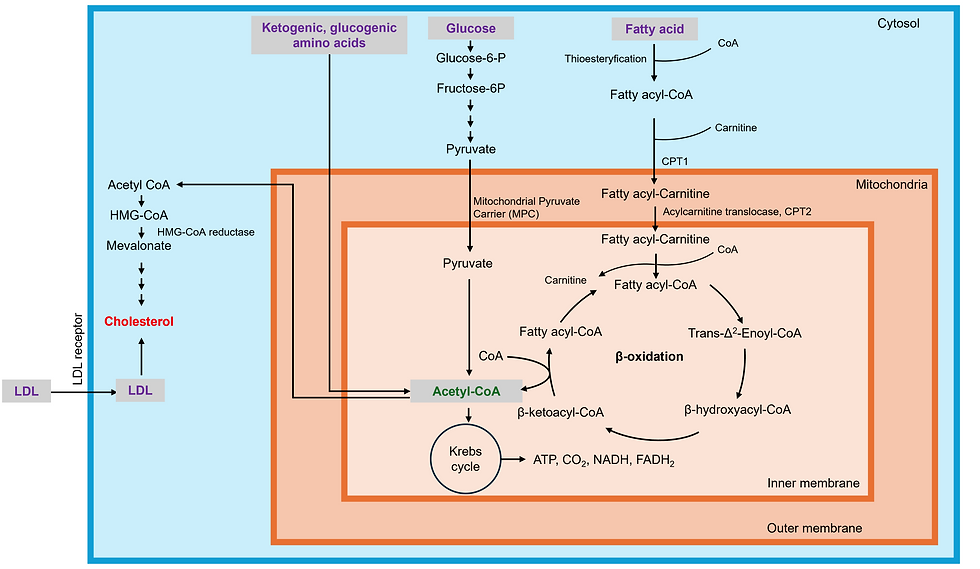
Cholesterol Synthesis: Converting Acetyl-CoA into Cholesterol
Cholesterol synthesis begins with acetyl-CoA, a molecule that serves as a precursor for many biosynthetic pathways, including the mevalonate pathway that leads to cholesterol production. Acetyl-CoA can be generated from various macronutrients, including fatty acids, glucose, and amino acids. Inside the liver and other tissues, acetyl-CoA enters the mevalonate pathway, where a series of enzymatic reactions eventually convert it into cholesterol. The most important regulatory step in this pathway is catalyzed by HMG-CoA reductase, an enzyme targeted by statins to reduce cholesterol levels.
Fatty Acids as a Source of Acetyl-CoA
Fatty acids are a major source of acetyl-CoA through the process of beta-oxidation. When dietary fats are broken down in the cells, they are converted into fatty acyl-CoA, which is transported into the mitochondria via the carnitine shuttle. Inside the mitochondria, fatty acids undergo beta-oxidation, which generates multiple molecules of acetyl-CoA, depending on the chain length of the fatty acid.
Glucose and Amino Acids Contributing to Acetyl-CoA Production
In addition to fatty acids, glucose serves as a critical source of acetyl-CoA. Glucose is first metabolized through glycolysis in the cytosol, producing pyruvate, which enters the mitochondria. There, pyruvate dehydrogenase converts pyruvate into acetyl-CoA, feeding into both energy production pathways and cholesterol synthesis. Certain amino acids can also be metabolized into acetyl-CoA. Ketogenic amino acids like leucine and lysine are converted directly into acetyl-CoA, while glucogenic amino acids are first converted into intermediates of the Krebs Cycle, or TCA cycle, before being funneled into acetyl-CoA production. This versatility allows cells to use a range of nutrients to support cholesterol synthesis, ensuring a steady supply of acetyl-CoA.
Exogenous Sources: Cholesterol from the Diet and LDL Uptake
While cells synthesizes a significant portion of its cholesterol, it can also acquire cholesterol from exogenous sources. Dietary cholesterol is absorbed in the intestines and packaged into chylomicrons, which transport lipids through the lymphatic system and into circulation. In addition, cells can obtain cholesterol from low-density lipoproteins (LDL), which carry cholesterol in the bloodstream. LDL receptors on the surface of cells bind LDL particles, allowing the cells to take up cholesterol through receptor-mediated endocytosis. This exogenous cholesterol is used to support membrane integrity, hormone production, and other cellular processes, balancing the cholesterol synthesized internally by the cell.
Balancing Cholesterol: Endogenous Synthesis and Exogenous Uptake
Cells carefully balances the levels of cholesterol from both endogenous and exogenous sources. When cholesterol levels are high, the synthesis of cholesterol is reduced, largely through the regulation of SREBP2, a transcription factor that controls the expression of key enzymes in cholesterol synthesis. Similarly, when cells have sufficient cholesterol, LDL receptor expression decreases, limiting the uptake of cholesterol from circulating LDL. This tight regulation helps maintain homeostasis, preventing excessive cholesterol accumulation, which can lead to the formation of atherosclerotic plaques and contribute to cardiovascular disease.
In summary, cholesterol synthesis begins with acetyl-CoA, which is derived from various nutrient sources including fatty acids, glucose, and certain amino acids. Cholesterol can also be sourced exogenously from the diet and through LDL uptake. Together, these processes ensure that cells have the cholesterol they need for critical functions, while mechanisms exist to prevent overaccumulation that can lead to disease.
6. Cholesterol homeostasis: Efflux, Synthesis, Uptake, Storage, and Trafficking

Cholesterol efflux is a key process that prevents cholesterol accumulation within cells, maintaining lipid homeostasis and protecting against diseases such as atherosclerosis. The primary pathway for cholesterol efflux is mediated by the ATP-binding cassette transporter A1 (ABCA1), which facilitates the transport of cholesterol to apolipoprotein A-I (ApoA-I). This process results in the formation of high-density lipoproteins (HDL), which carry cholesterol to the liver for excretion. The activity of ABCA1 is regulated by Liver X Receptor (LXR), a nuclear receptor activated by oxysterols, which are oxidized derivatives of cholesterol produced when cellular cholesterol levels rise. Once activated, LXR upregulates ABCA1 expression, promoting cholesterol export from cells and preventing cholesterol buildup. Additionally, ABCG1 plays a complementary role by exporting cholesterol to mature HDL particles, further contributing to cholesterol efflux, particularly in cells like macrophages.
When cholesterol homeostasis is disrupted, efflux pathways become impaired, leading to cholesterol accumulation within cells. This can result from reduced LXR activity or decreased expression of efflux transporters like ABCA1, often caused by inflammatory conditions. Chronic inflammation, triggered by excess cholesterol, further disrupts these pathways. One of the most important mechanisms linking cholesterol accumulation to inflammation is NLRP3 inflammasome activation, which is triggered by cholesterol crystal formation. Cholesterol crystals physically disrupt lysosomes, leading to the activation of the NLRP3 inflammasome and the release of pro-inflammatory cytokines like IL-1β. IL-1β amplifies the inflammatory response and activates the NF-κB pathway, which in turn suppresses LXR activity and reduces ABCA1 expression, creating a vicious cycle of cholesterol accumulation and inflammation.
A key factor contributing to the disruption of cholesterol efflux is the overactivation of SREBP-2 (Sterol Regulatory Element-Binding Protein 2). SREBP-2 is a transcription factor that controls cholesterol synthesis and uptake by upregulating the expression of HMG-CoA reductase (HMGCR), the rate-limiting enzyme in cholesterol biosynthesis. Importantly, HMGCR is the target of statins, a class of drugs that lower cholesterol by inhibiting this enzyme. This process predominantly occurs in the liver, the main organ responsible for cholesterol synthesis. SREBP-2 also promotes the expression of the LDL receptor (LDLR), which facilitates cholesterol uptake from low-density lipoproteins (LDL). Excessive SREBP-2 activation, which can be driven by mTORC1 activation, leads to overproduction of cholesterol, overwhelming the cell's capacity for efflux and resulting in intracellular cholesterol accumulation.
mTORC1 activation not only increases cholesterol synthesis but also plays a role in autophagy dysfunction, which contributes to cholesterol accumulation. Autophagy is a critical process for degrading and recycling cellular components, including lipids and damaged organelles. When mTORC1 is overactivated, it inhibits autophagy, impairing cellular processes like mitophagy (the degradation of damaged mitochondria) and lipophagy (the degradation of lipid droplets). This disruption in autophagy results in the accumulation of damaged mitochondria and lipid droplets, further promoting cholesterol buildup and exacerbating cellular dysfunction.
A key consequence of autophagy dysfunction is mitochondrial dysfunction, which significantly contributes to ROS production. When mitophagy is inhibited due to autophagy dysfunction, damaged mitochondria accumulate, leading to inefficient electron transport and excess ROS production. ROS causes oxidative damage to lipids, proteins, and DNA, exacerbating cellular dysfunction. This oxidative stress activates NF-κB signaling, which not only drives a sustained pro-inflammatory response but also suppresses LXR activity, further impairing cholesterol efflux through reduced ABCA1 expression. Additionally, the failure to clear lipid droplets via lipophagy worsens intracellular lipid accumulation, reinforcing the cycle of mitochondrial damage, ROS production, and inflammation. In this pro-inflammatory environment, oxidized LDL (oxLDL) can activate TLR4, further promoting inflammation and perpetuating cholesterol accumulation.
To avoid toxicity from free cholesterol accumulation in cellular membranes, excess cholesterol is stored in its esterified form as cholesteryl esters. The enzyme responsible for this process is acyl-coenzyme A: cholesterol acyltransferase (ACAT), which is found in the endoplasmic reticulum (ER). ACAT catalyzes the esterification of cholesterol by attaching a fatty acid from fatty acyl-CoA to cholesterol, producing cholesteryl esters. These esters are stored in lipid droplets within the cell, particularly in the liver and adipose tissue. Cholesterol esterification helps regulate free cholesterol levels in the plasma membrane and other cellular compartments. Under certain conditions, such as in foam cell formation in atherosclerosis, cholesteryl esters can accumulate excessively, contributing to disease progression. When needed, cholesteryl esters are hydrolyzed by cholesteryl ester hydrolases (CEH) to release free cholesterol for membrane synthesis or steroid hormone production.
Intracellular trafficking also plays a critical role in maintaining cholesterol homeostasis. Cholesterol is synthesized in the endoplasmic reticulum (ER) and then transported to various cellular compartments such as the plasma membrane, mitochondria, and lysosomes. Specialized lipid transport proteins, such as Niemann-Pick C1 (NPC1) and Niemann-Pick C2 (NPC2), facilitate the movement of cholesterol out of lysosomes after the degradation of low-density lipoprotein (LDL). When these transport mechanisms are impaired, such as in Niemann-Pick disease, cholesterol accumulates in lysosomes, leading to cellular dysfunction. Additionally, cholesterol is transferred between the ER and plasma membrane through vesicular transport and non-vesicular routes, involving proteins like StAR-related lipid transfer (START) domains and oxysterol-binding proteins (OSBPs). This trafficking network ensures that cholesterol is distributed where it is needed, particularly in the plasma membrane where it maintains membrane fluidity, or in mitochondria where it is involved in steroidogenesis. Disruptions in this trafficking system can contribute to cholesterol accumulation in specific organelles, exacerbating metabolic dysfunction and disease.
In conclusion, maintaining cholesterol homeostasis requires a careful balance between efflux, synthesis, uptake, storage, and intracellular trafficking. Efflux pathways, mediated by transporters like ABCA1 and ABCG1 under the control of LXR, prevent excess cholesterol buildup, while SREBP-2 regulates cholesterol synthesis and uptake, ensuring cells acquire sufficient cholesterol when needed. To prevent toxicity from free cholesterol, the enzyme ACAT esterifies excess cholesterol, storing it as cholesteryl esters in lipid droplets, which can be mobilized when required for membrane synthesis or hormone production. Disruptions in these processes, such as impaired efflux, overactivation of cholesterol synthesis, or defective cholesterol storage and trafficking, can lead to cholesterol accumulation in specific organelles, including lysosomes and the plasma membrane. This imbalance is further exacerbated by inflammation, mitochondrial dysfunction, and autophagy impairment, all of which contribute to the accumulation of cholesterol and cholesteryl esters. The formation of cholesterol crystals activates the NLRP3 inflammasome, while oxidized cholesterol and ROS promote inflammation through pathways like TLR4, creating a cycle of inflammation and lipid overload. Understanding the complex interplay between these mechanisms—efflux, synthesis, storage, trafficking, and inflammatory responses—is essential for developing targeted therapies to prevent or reverse cholesterol-related diseases.